This article was published as a part of the Data Science Blogathon.
Introduction
In this article, I will attempt to explain all of the ideas that you should be familiar with about databases. As we all know, while working on a Data Science, Machine Learning, Deep Learning, or another project, the most important element is the data, which is considered to be the fuel for all of the Machine or Deep Learning algorithms.
So, while working on real-time projects, we have to focus upon our database, and we have to check it is centralized or not, if it includes corrupt data or not, because corrupted data may cause redundancy in your entire database also it can give the inconsistent results when we put input user query, etc.
So, knowing all these basics of a database becomes critical before studying Data Science, and also helpful to become a Data Scientist or Machine Learning Engineer because these are standard things and can also be asked in interviews. 🥳
Table of Contents
1. What is meant by the term “Database”?
2. What are Database Management systems?
3. Need and Use of Entity-relationship diagram
4. Entity Set and its types – weak and strong entity set
5. Relationship, Cardinality Constraints
6. Meaning of attributes and their types
7. While creating a relational database model, what constraints do we have to follow
8. Concept of Key with their types
9. Why do we need normalization and learning different normal forms?
What is a Database?
The database is defined as the actual connections or a connection of information that has some semantic association. I used the term “semantic” in this sentence; are you familiar with it?
Semantic means that there is a logical relationship in the database. As a result, by putting the meaning of the term semantic in the above-mentioned definition, the enlarged definition of the database becomes,
Definition: A database is a combination of linked data that represents a real-world situation. A database system is designed to be created and populated with data in order to complete a specific purpose.
In the above-mentioned definition, I’ve used the word Database system, which is sometimes used interchangeably with the term Database Management system. So, please learn about both of these concepts and if you feel that both these terms are not different, then let me know in the Comment Box. 🤔
Database Management Systems
Database Management systems (DBMS): It is software for storing and retrieving user data while focusing on the proper security. It is made up of a combination of applications that alter the database. The DBMS takes the data from applications and tells the operating system to give the data which the user wants. A database management system (DBMS) assists users and other third-party applications in storing and retrieving data in real-time systems.
Database management systems were created to address the following issues with traditional file-processing systems that are supported by traditional operating systems:
– Data redundancy and inconsistency
– Difficulty in accessing data
– Data isolation – multiple files and formats
– Integrity problems
– Atomicity of updates
– Concurrent access by multiple users
– Security problems
Entity-Relationship Diagram
The Entity-Relationship Diagram (ER diagram) is a conceptual model that depicts the logical structure of the database in a graphical manner. It displays all of the constraints and relationships that exist between the different components of the databases. The following three elements make up a given ER diagram:
Entity Sets, Attributes, and Relationship Sets.
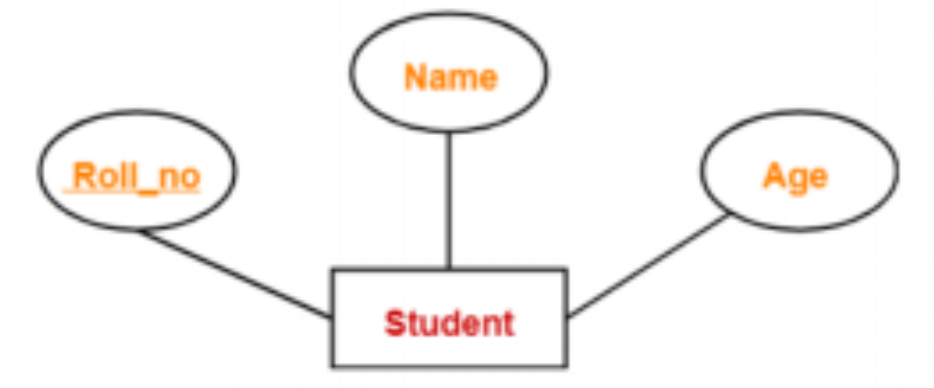
Here the primary key is Roll no, which can be used to uniquely identify each entity. As a result, a student’s roll number can be used to identify them individually.
Entity Set
An entity set is a set of the same type of entities.
Strong Entity Set:
A strong entity set is one that has enough properties to uniquely identify all of its entities. In other words, there is a primary key for a strong entity set.
– A strong entity set’s primary key is expressed by underlining it.
Weak Entity Set:
A weak entity set is one that lacks sufficient properties to uniquely identify its entities.
– In other words, there is no primary key for a weak entity set. It does, however, include a partial key, which is called a discriminator.
– Discriminator is capable of identifying a subset of entities in the complete set of entities. The discriminator is denoted by a dashed line underlined.
Relationship
A relationship is a connection between two or more entities.
Unary Relationship Set: An unary relationship set is a relationship set in which only one entity set participates.
Binary Relationship Set: A binary relationship set is a relationship set in which two entity sets from the complete set participates.
Ternary Relationship Set: A ternary relationship set is a relationship set in which three entity sets from the complete set participates.
N-ary Relationship Set: An N-ary relationship set is a relationship set in which ‘n’ entity sets participate.
Cardinality Constraint
The maximum number of related components in which an entity can participate is defined by the cardinality constraint.
One-to-One Cardinality: An entity in set A can only be related to one entity in set B. An entity in set B can only be linked to one entity in set A.
One-to-Many Cardinality: Any number (zero or more) of entities in set A can be related to any value (zero or more) of entities in set B. An entity in set B can only be linked to one entity in set A.
Many-to-One Cardinality: An entity in set A can only be related to one entity in set B. An entity from set B can be linked to any quantity of entities from set A.
Many-to-Many Cardinality: An entity in set A can be related to any number of entities in set B (zero or more). An entity in set B can be linked to any quantity of entities in set A (zero or more).
Attributes
Attributes are the descriptive qualities that each entity in an Entity Set possesses. The following are the different types of Attributes:
Simple Attributes: Simple attributes are ones that cannot be further subdivided. Age is an example.
Composite Attributes: Composite traits are ones that are made up of simple attributes. for example, Name and address.
Multi-Valued Attributes: Multi-valued attributes are those that can have several values for a particular entity in an entity set. For example, mobile phone number and email address.
Derived Attributes: These are attributes that may be derived from other attributes (s). Age, for example, age can be calculated using the date of birth (DOB).
Key Attributes: Key attributes are those that may uniquely identify an entity in an entity collection. As an example, Roll No.
Constraints
Relational constraints are limitations placed on database contents and processes. This implies that the data in the database is consistent.
Domain Constraint: This constraint defines the set of values for an attribute. It states that the attribute’s value must be chosen from its set of values.
Tuple Uniqueness Constraint: The tuple uniqueness constraint states that all tuples in any relationship must be necessarily unique.
Key Constraints: The primary key’s values must all be unique and also the primary key value cannot be null.
Entity Integrity Constraint: The entity integrity constraint states that no attribute of the main key in any relationship may have a null value.
Referential Integrity Constraint: This constraint states that any values assigned to the foreign key must be either available in the relation of the primary key or null.
Keys
A key is a set of attributes that may uniquely identify each tuple in a given relation. Key Varieties:
Super key: A super key is a set of attributes that may uniquely identify each tuple in a given relation. A super key can contain any number of attributes.
Candidate key: A candidate key is a set of the minimum attribute(s) that may uniquely identify each tuple in a given relation.
Primary Key: A primary key is a candidate key chosen by the database designer while constructing the database. Primary keys are one-of-a-kind and NOT NULL.
Alternate key: Alternate keys are candidate keys that are left unimplemented or unused once the primary key is implemented.
Foreign Key: When the values of one attribute ‘X’ are dependent on the values of another attribute ‘Y,’ the attribute ‘X’ is said to be a foreign key to that other attribute ‘Y.’ Here the relation that contains the attribute ‘Y’ is known as the referred relation. The referring connection is the one in which the attribute ‘X’ exists.
Composite Key: A composite key is a primary key that is made up of numerous qualities rather than simply one.
Unique Key: It is unique for all of the table’s records. Its value cannot be modified once it has been allocated; it is non-updatable. It might have a value of NULL.
Normalization
Database normalization helps in making the database consistent by-
– Eliminating redundancies
– Data integrity is ensured by lossless decomposition.
Types of Normal forms:
First Normal Form (1NF): A given relation is said to be in First Normal Form (1NF) if each cell in the table has just one atomic value, i.e. if every tuple’s attribute is either single-valued or null.
Second Normal Form (2NF): A given relation is said to be in Second Normal Form (2NF) if and only if the following conditions are met.
– The relationship is already present in 1NF.
– There is no partial dependency in the relationship.
A partial dependency, defined from A to B if and only if A is a subset of some candidate key and B is a non-prime attribute.
Third Normal Form (3NF): A given relation is said to be in Third Normal Form (3NF) if and only if the following conditions are met:
– In 2NF, a relationship already exists.
– There is no transitive reliance on non-prime characteristics.
A transitive dependence exists if and only if A is not a super key and B is a non-prime attribute.
BCNF (Boyce-Codd Normal Form): In BCNF, a given relation is termed iff:
– The relationship is already present in 3NF.
– For any non-trivial functional dependence ‘A->B,’ A is the relation’s super key.
This completes all the basics of databases which are very important while we are going to learn SQL or there are also some concepts like Query Optimization and processing, these are higher-level databases concepts.
Other Blog Posts by Me
If you want to read my previous blogs, you can read from here.
Previous Data Science Blog posts.
Here is my Linkedin profile if want to connect with me.
End Notes
Thanks for reading!
I hope that you have enjoyed the article. If you like it, share it with your friends also. Something not mentioned or want to share your thoughts? Feel free to comment below And I’ll get back to you. 😉
I am a B.Tech. student (Computer Science major) currently in the pre-final year of my undergrad. My interest lies in the field of Data Science and Machine Learning. I have been pursuing this interest and am eager to work more in these directions. I feel proud to share that I am one of the best students in my class who has a desire to learn many new things in my field.